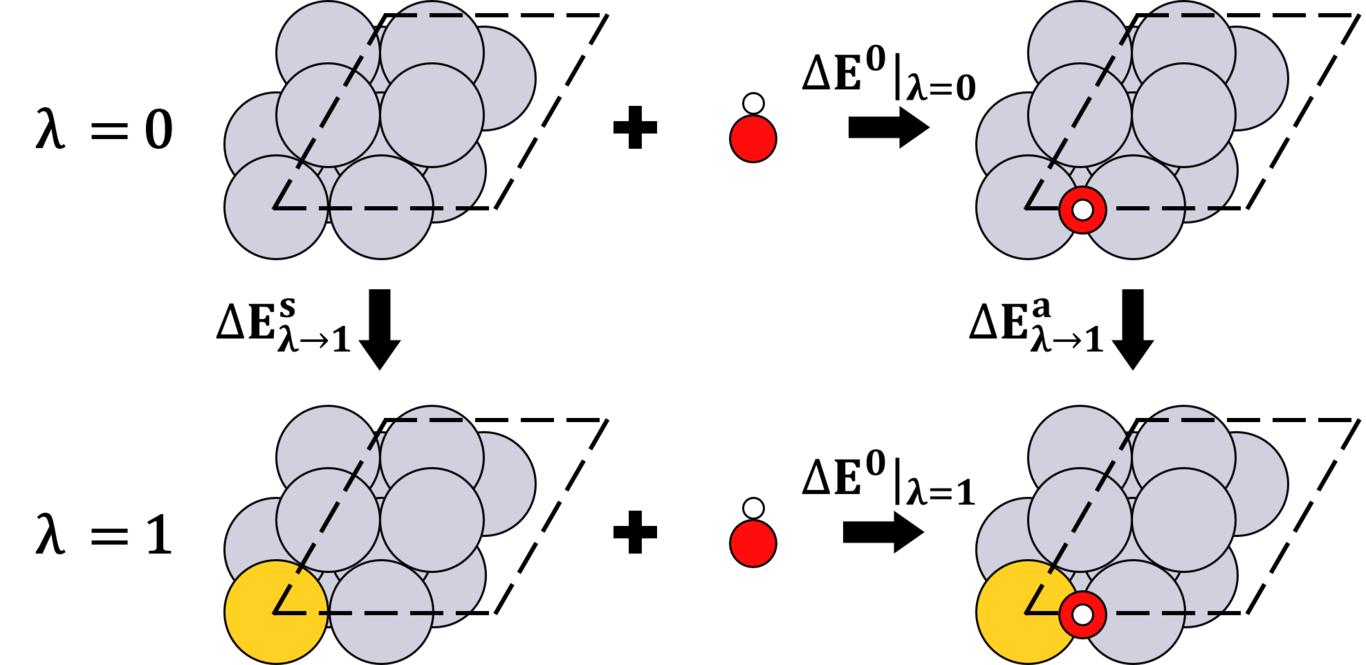
Reviewer 1's original text is given in bold while our responses are in plain text. 1. My main question is whether the authors have seen any kind of finite-size effect on the alchemical potential due to the minimal cell employed in this study. Due to the coulombic form of the alchemical potential, it appears unlikely that there is no such effect, but potentially it is a quite systematic contribution for all target systems considered. While the errors might be attributed as due to finite-size effects, our group has considered smaller and larger unit cells to describe adsorbates bound to the surface with different coverages. We find that first order APDFT is generally more accurate when alchemical derivatives of or near the transmuted atoms are low, and less accurate when alchemical derivatives of or near the transmuted atoms are high. For small unit cells with high adsorbate coverages, errors are usually high, but for large unit cells they are still high if the transmuted atom is near the binding site of the adsorbate. For now, we prefer to think of errors in terms of the magnitude of the alchemical derivatives near the transmuted sites, though considering this as a finite-size effect may not be incorrect. 2. The authors state “Computational alchemy is a perturbation theory approximation”. It might be warranted to rather emphasise in the article that it is the Taylor series truncation that renders it an approximation. Under the supposition that the Taylor series converges, in fact the expansion should be exact. Admittedly, not all of the terms (e.g. basis function changes in the direction of the alchemical change) are typically taken into account explicitly, but only if the perturbation approach is exact in the first place, computational alchemy can expect to have the predictive power that is required for the materials design applications outlined in this work. This is an excellent point, and we have revised the text here as well as changed most mentions of "computational alchemy" to be "first order APDFT" as the latter is more precise along these terms. We have also changed the text explaining Eq. 4 to better clarify. 3. Similarly, when the authors discuss the charge neutrality of the alchemical change, it might be important to highlight that the theory does not require the changes in nuclear charges to be strictly compensated. In this particular application however, the change should be neutral as otherwise the total surface charge density of the interface becomes unphysically large. We have revised our text to highlight this point. 4. When discussing the energy barriers relative to DFT data, the authors give an error bound of 0.3eV. To set this into perspective: how accurate can the DFT method expected to be for the systems at hand? Effectively, computational alchemy constitutes a Taylor expansion on the potential energy surface of the reference method used, therefore can only hope to recover the accuracy of the reference method. If this error bound is low compared to DFT vs higher level methods, this would effectively render computational alchemy to have predictive power indistinguishable from the reference method. This is a good point, but we prefer making a few more qualifying statements. The text has now been changed to be:"Even though first order APDFT can exhibit errors as large as 0.3 eV in barrier heights for reference system doped with just a single atom, it is promising that such simple approximations can be useful to calculate a computationally expensive descriptor that guides screening studies. Furthermore, as the source for errors in different APDFT approximations become better understood, it becomes reasonable to imagine that more accurate approaches might be developed based on APDFT that would have a predictive power comparable (or even indistinguishable) to standard DFT."5. In Figure 5, the authors show the NEB paths found from computational alchemy. For the case of Delta Z=-1, NEB image 7 has a substantially lower energy, even lower than the endpoint of the NEB path. What is the reason for this given that alchemical predictions are done on all NEB images in the same fashion? We apologize that this was not clear. The energies reflect a change in barrier height with respect to that calculated for the reference system. Thus, we are not reporting a negative barrier, but a barrier that is lower than the reference calculation. Text has been clarified in the caption. 6. Figure 5 b) is hard to assess visually, since most of the data points are obscured by others. Perhaps a plot of signed difference alchemy-DFT vs distance from the transmutation site would be clearer. Moreoever, no units are given. This is a good suggestion, but to be consistent with other data showing parity plots in this work as well as previous work, we will keep the data as shown. The main point of the figure is simply to show that this approach is accurate when In future work we will focus more on elucidating errors and how to correct them, and this suggestion would be very helpful. We now mention the units in the captions . 7. In the caption of Figure 2, some cross-references seem to be mixed up. We have corrected these cross references. We thank Reviewer 1 for reading our manuscript and providing helpful suggestions to improve our manuscript. We also thank Reviewer 2 for making comments within the Authorea document about typos. We believe we have addressed all points, and we hope that the manuscript can now be considered suitable for publication in Int. J. Quantum Chem.